The sbmlpbk package
Johannes Kruisselbrink
2025-10-26
sbmlpbk.RmdThe sbmlpbk package
An R package for importing Physiologically Based Kinetic (PBK) models
encoded in the Systems Biology Markup Language (SBML) format, enabling
simulation using the deSolve package.
Loading a model
Use the function load_sbml to load a PBK model from an
SBML file. As an example, the package includes a simple SBML PBK model
simple_oral.sbml. The following example shows how to load
the model from this file:
## Load SBML model from file
file_simple_oral <- system.file("extdata/", "simple_oral.sbml", package = "sbmlpbk")
model <- load_sbml(file_simple_oral)The output of the load_sbml function is an object of
class sbmlModel. This object is a named list
with of the following elements:
- compartment: A named list with the IDs and default sizes of the model compartments.
- species: A named list with the IDs and other info of the model species.
- param: A named list with the IDs and (default) values of the model parameters.
- function_defs: A list of the additional functions defined in the SBML file, each in the form of character strings representing R expressions.
- reactions: A list of reactions, each defined as a list of ID, reactant (species ID), product (species ID), and kinetic law (character string of R expression).
- assignment_rules: A list of the assignment rules defined in the SBML file, each in the form of character strings representing the R expression.
- rate_rules: A list of the rate rules defined in the SBML file, each in the form of character strings representing the R expression.
- odes: A list of ODEs generated from the SBML file, each in the form of character strings representing the R expression.
- default_params: Vector with the default values of the parameters of the model.
-
func: A
deSolvecompliant R-function for running simulations using theodefunction.
The summary function can be used to get a short
description of the content of an sbmlModel object.
summary(model)
#> ==================
#> SBML Model Summary
#> ==================
#>
#> Compartments: 5 [ Gut, Blood, Liver, Rest, Urine ]
#> Species: 5 [ AGut, ABlood, ALiver, ARest, AUrine ]
#> Parameters: 19 [ BW, VRc, VLc, VBc, QCC, ... ]
#> Functions: 0
#> Assignment rules: 6
#> Rate rules: 0
#> Reactions: 6
#>
#> Time unit: h
#> Amount unit: ug
#> Volume unit: LInspecting model equations
To print the equations of the loaded model, type:
print(model, type = 'equations')
#>
#> # --- Assignment rules ---
#> Blood <- BW * VBc
#> Liver <- BW * VLc
#> Rest <- BW * VRc
#> QC <- BW^0.75 * QCC
#> QL <- QC * QLc
#> QR <- QC * QRc
#>
#>
#> # --- Transfer ODEs ---
#> dAGut <- -AGut * Ka
#> dABlood <- -ABlood * QL/Blood + ALiver * QL/(Liver * PCLiver) - ABlood * QR/Blood + ARest * QR/(PCRest * Rest) - ABlood * CLUrine/Blood
#> dALiver <- AGut * Ka + ABlood * QL/Blood - ALiver * QL/(Liver * PCLiver)
#> dARest <- ABlood * QR/Blood - ARest * QR/(PCRest * Rest)
#> dAUrine <- ABlood * CLUrine/BloodRunning simulations
The code below shows how to run simulations using the
ode function of deSolve.
## Load model functions
load_functions(model)
## Load params
params_csv <- system.file("extdata/", "simple_oral_params.csv", package = "sbmlpbk")
params <- load_params(model, file = params_csv, param_instance = "simple_PARAM")
# Set input species
input_species <- "AGut"
# Set input events (single unit bolus at time 5)
eventdat <- data.frame(var = c(input_species), time = c(5), value = c(1), method = c("add"))
# Set simulation times
times <- seq(0, 40, 0.1)
# Set initial states
initial_states <- setNames(rep(0, length(model$species)), names(model$species))
# Load deSolve model function
func <- create_desolve_func(model)
# Simulate
out <- ode(
y = initial_states,
times = times,
func = func,
parms = params,
events = list(data = eventdat)
)In this example, the function load_params loads the
parameter values from a CSV file. Alternatively,
model$default_params can be passed to use the model’s
default parameters. The function load_functions loads the
additional functions defined in the SBML file. The function
create_desolve_func creates the function that can be used
as func argument of deSolve’s ode
function.
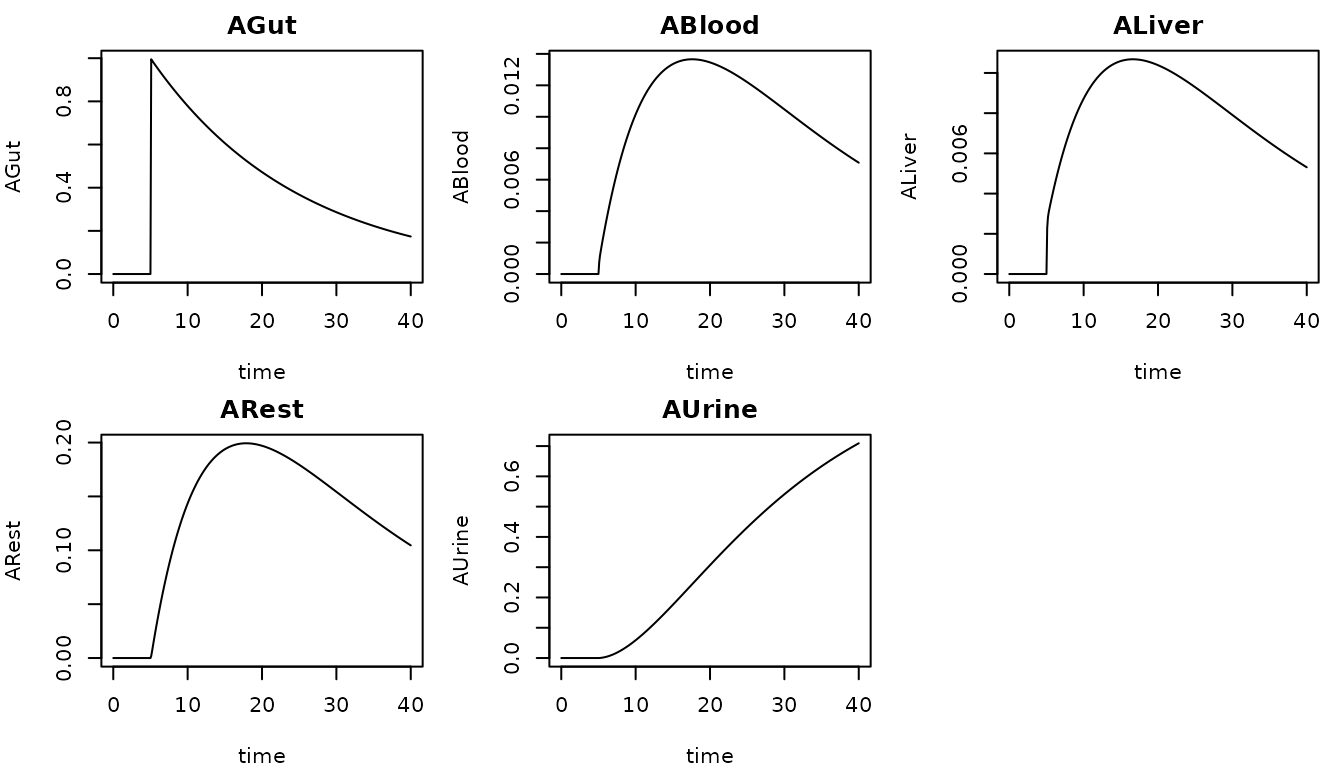
From here, the simulation output is ready for any further processing. We can, for example, plot the output time series as follows:
# Set up plotting layout
n_cols <- 3
n_rows <- ceiling(length(model$species) / n_cols)
par(mfrow = c(n_rows, n_cols), mar = c(4, 4, 2, 1))
# Loop through variables and plot
for (species in names(model$species)) {
plot(out[,1], out[,species],
type = "l",
main = species,
xlab = "time",
ylab = species)
}
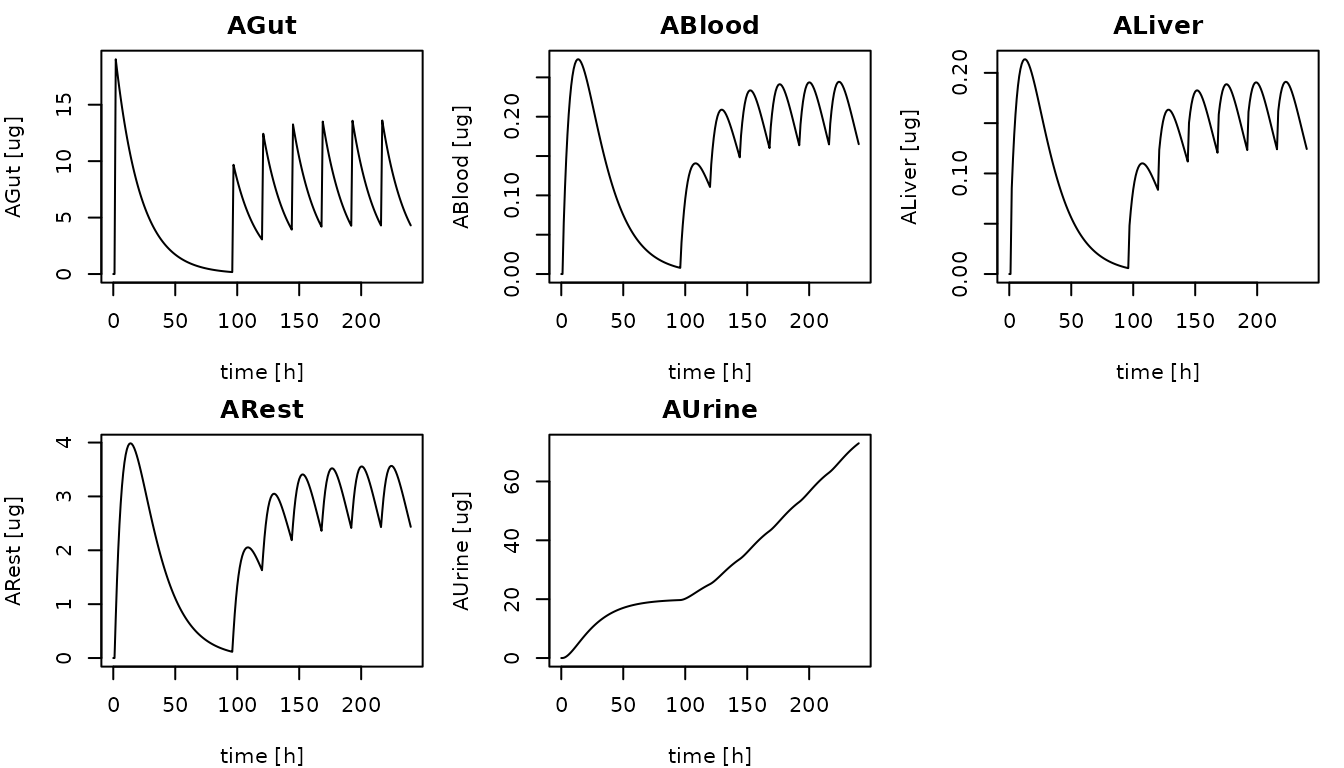
Running dosing scenarions
Running PBK model simulations with various dosing scenarios requires
construction of deSolve patterns. As PBK models may differ
in time resolution (e.g., hours/days) and dosing amounts (e.g., mg, ug,
moles) creation of dosing scenarios, running simulations, and evaluating
outputs requires alignment of the model’s time resolution and
dosing/output amounts with the desired resolutions. The methods and are
provided to assist with this. The method creates
deSolve-compatible events from a specified dosing scenario
in a user-specified time unit and amounts unit. Likewise, the method
creates a compliant sequence, specifying the times for which simulation
output is wanted.
dosing_events <- list(
# Single bolus dose of amount 20 at time 1 (in days)
list(
target = "AGut",
dose_type = "single_bolus",
time = 1,
amount = 20
),
# Repeated bolus dose with amount 10, starting at day 4, repeating
# every day until time day 10
list(
target = "AGut",
dose_type = "repeated_bolus",
time = 4,
amount = 10,
interval = 1,
until = 10
)
)
# Set simulation times; 10 days with hourly evaluation resolution
num_days = 10
evals_per_day = 24
times <- create_desolve_times(
model,
duration = num_days,
step = 1/evals_per_day,
unit = 'd'
)
# Create and set deSolve dosing events
eventdat <- create_desolve_events(
model,
dosing_events,
time_unit = 'd', # events time unit was in days
amount_unit = 'ug'
)
# Set initial states
initial_states <- setNames(rep(0, length(model$species)), names(model$species))
# Load deSolve model function
func <- create_desolve_func(model)
# Simulate
out <- ode(
y = initial_states,
times = times,
func = func,
parms = model$params,
events = list(data = eventdat)
)
# Set up plotting layout
n_cols <- 3
n_rows <- ceiling(length(model$species) / n_cols)
par(mfrow = c(n_rows, n_cols), mar = c(4, 4, 2, 1))
# Loop through species and plot
for (species in names(model$species)) {
plot(out[,1], out[,species],
type = "l",
main = species,
xlab = paste("time [", summary(model)$time_unit, "]", sep=""),
ylab = paste(species, " [", summary(model)$amount_unit, "]", sep=""))
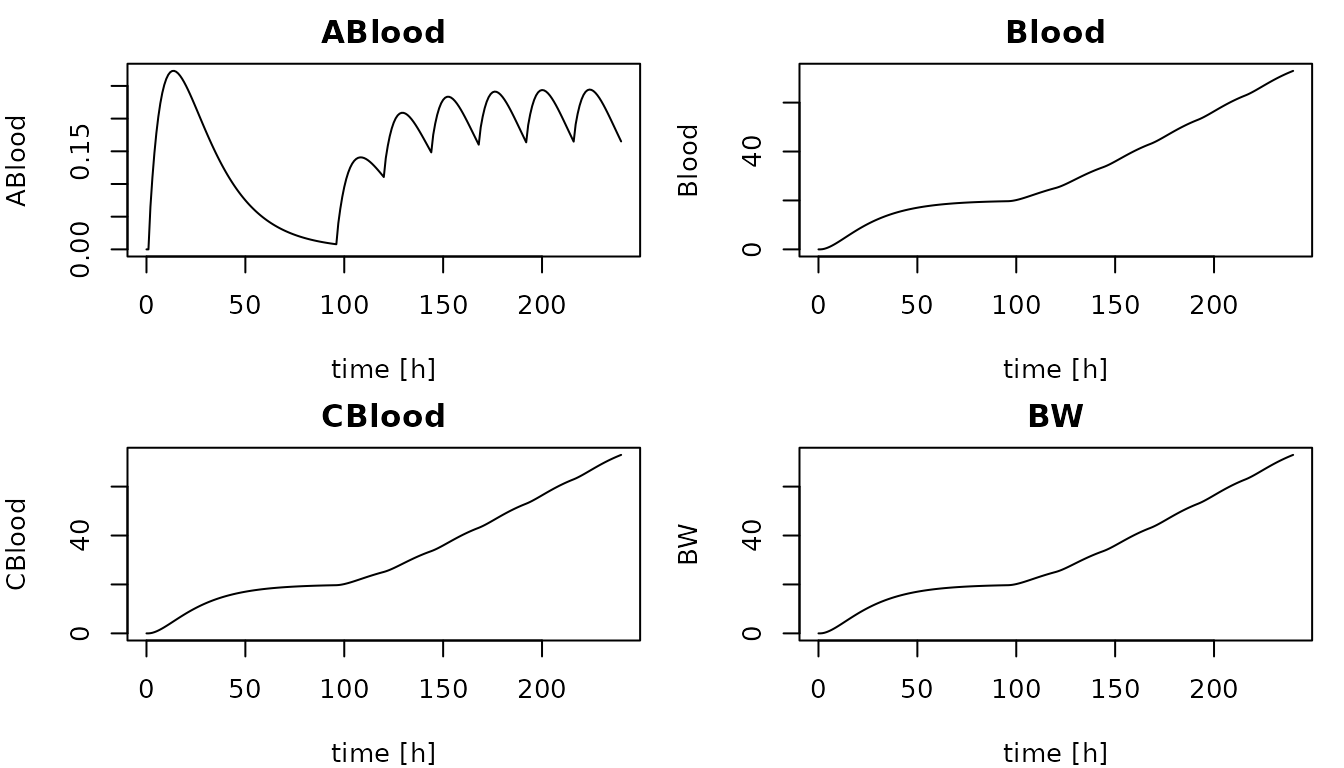
} ## Additional simulation outputs
## Additional simulation outputs
Additional simulation outputs, such as dynamic parameters,
compartment sizes, but also outputs as concentrations, can be included
via the outputs argument of the
create_desolve_func function. In the code below, three
additional outputs are defined. Size of the blood compartment (i.e.,
blood volume), concentration in blood, and the value of the body weight
parameter (here constant during simulation).
# Define additional outputs
outputs <- list(
Blood = "Blood",
CBlood = "ABlood / Blood",
BW = "BW"
)
# Load deSolve model function with additional outputs
func <- create_desolve_func(
model,
outputs = outputs
)
# Simulate
out <- ode(
y = initial_states,
times = times,
func = func,
parms = model$params,
events = list(data = eventdat)
)
# Set up plotting layout
par(mfrow = c(2, 2), mar = c(4, 4, 2, 1))
# Plot amount of chemical in blood
plot(out[,1], out[,'ABlood'],
type = "l",
main = 'ABlood',
xlab = paste("time [", summary(model)$time_unit, "]", sep=""),
ylab = 'ABlood')
# Loop through outputs and plot
for (output in names(outputs)) {
plot(out[,1], out[,species],
type = "l",
main = output,
xlab = paste("time [", summary(model)$time_unit, "]", sep=""),
ylab = output)
}