Introduction to the statgenGWAS package
Bart-Jan van Rossum
2026-02-19
Source:vignettes/GWAS.Rmd
GWAS.RmdThe statgenGWAS package
The statgenGWAS package has been designed for performing single trait Genome Wide Association Studies (GWAS), where the objective is to find associations between genetic markers and phenotypic traits.
Theoretical background
Suppose there are n measurements of
a phenotype for t genotypes. Then a
very common linear mixed model for association mapping is y = X\beta + Zu + e,
where y is an n \times 1 vector of observed phenotypes and
X is an n
\times q design matrix for the fixed effects, including an
intercept, covariates, and the SNP of interest. \beta is the q
\times 1 vector of coefficients of these fixed effects, the last
element (\beta_{\textrm{SNP}}) being
the SNP-effect of interest. Z is the
n \times t incidence matrix assigning
individuals to genotypes. u is the
vector of genetic random effects, with var(u)
= \sigma^2_g K, where K is a
marker-based relatedness matrix. Finally, e is a n \times
1 vector of residual (non-genetic) errors, with var(\epsilon) = \sigma^2_e I_n. For this
model the phenotypic variance-covariance matrix is \Sigma = \sigma^2_g ZKZ^{t} + \sigma^2_e
I_n.
Application in the package
Single trait GWAS in the statgenGWAS package follows the approach of Kang et al. (2010) and related papers:
- For the ‘empty’ model (i.e., without any SNP in the fixed effects), we obtain REML-estimates of the variance components \sigma^2_g and \sigma^2_e, which give an estimate \hat \Sigma of the variance-covariance matrix.
- Fixing \hat \Sigma, we use generalized least-squares (GLS) to test the null-hypothesis \beta_{\textrm{SNP}} = 0 using an F-test, for each SNP in turn. The last step gives vectors of estimated effect-size and P-values for all SNPs.
These steps are repeated for each combination of trial and trait for which the analysis is run.
The REML-estimates in step (1) can be computed using either the Efficient Mixed Model Association (EMMA) algorithm (Kang et al. 2008), or the Newton-Raphson (NR) algorithm (Tunnicliffe 1989). Our implementation of GLS is a c++ implementation of the R code of Segura et al. (2012).
The class gData
Genotypic data are stored in an object of class
gData (genotypic
Data). A gData object will contain all data needed for
performing analyses, so the first thing to do when using the statgenGWAS
package is creating a gData object. This can be done using the function
createGData. A gData object combines genotypic
data and phenotypic data in a list. In a typical workflow
the user first imports marker and map data as illustrated in the
following example.
If an existing gData object is provided as an input to the
createGData() function, data is added to the existing
object and existing data will be overwritten with a warning if
necessary. All data is checked for consistency and, if needed, default
names are added with a warning message.
A gData object created this way will consist of the following
components:
Marker map
The data.frame map is used to describe the physical positions of the markers on the chromosomes. The data consists of two columns, chr for the name or number of the chromosome and pos for the position of the marker on the chromosome. The position can be in basepair or in centimorgan. The row names of map should be the names of the markers.
Marker matrix
The marker matrix is stored in the matrix marker within the gData object. The column names should be the names of the markers and the row names the names of the genotypes. Markers can either be coded as character strings or as numerical values. In the former case, before performing any analysis, the marker matrix has to be converted to a numerical matrix. This can be done using the function codeMarkers.
Phenotypic data
Phenotypic data, either directly from field trials or after summarizing can be stored in pheno in the gData object. Pheno can either be a single data.frame or a list of data.frames for storing data for different trials or different summarizations of the original data. The first column of all elements of pheno should be genotype and all the other columns should represent different traits. Storing additional variables should be done in covar. A single data.frame will be converted to a one-item list upon creation of a gData object.
Kinship matrix
The kinship matrix describes the genetic relatedness between the different genotypes. It is stored in the matrix kinship in the gData object. This should be a square matrix with genotypes in both row and column names and a measure for the genetic relatedness in its cells. There are many ways of computing genetic relatedness some of which are included in this package. Instead of providing one kinship matrix a list of kinship matrices can be provided with each matrix in this list a chromosome specific kinship matrix, usually computed using all markers that are not on the corresponding chromosome. If a named list of kinship matrices is provided, its names should match the names or numbers of the chromosomes in map. Unnamed lists will be given default names.
Further covariates
Further covariates that can be used in the analyses can be stored in the data.frame covar. This data.frame has genotypes in its row names and the covariates in the column names. Covariates can be numerical, factor or character data. The latter will be converted to a factor when creating the gData object.
Worked example
As an example of the functionality of the package a worked example is provided using maize data from the European Union project DROPS. The data is available from https://entrepot.recherche.data.gouv.fr/dataset.xhtml?persistentId=doi:10.15454/IASSTN (E. J. Millet et al. 2019) and the relevant data sets are included as data.frames in the statgenGWAS package.
Load data
The first step is loading the data into R.
dropsMarkers contains the coded marker information for 41722 SNPs and
246 genotypes. dropsMap contains information about the positions of
those SNPs on the B73 reference genome V2. dropsPheno contains data for
the genotypic means (Best Linear Unbiased Estimators, BLUEs), with one
value per experiment per genotype, for eight traits. For a more detailed
description of the contents of the data see
help(dropsData).
Create gData object
The next step is to create a gData object from the raw data that can be used for the GWAS analysis. For this the raw data has to be converted to a suitable format for a gData object. When creating a gData object, the markers should be input as a matrix or data.frame with genotypes in the row names and markers in the column names. The latter is already satisfied so only the row names should be added and the Ind column used for that should be dropped.
## Add genotypes as row names of dropsMarkers and drop Ind column.
rownames(dropsMarkers) <- dropsMarkers[["Ind"]]
dropsMarkers <- dropsMarkers[colnames(dropsMarkers) != "Ind"]The map used as input when creating a gData object, should at least have the columns “chr” and “pos” and the row names should be the names of the SNPs. Other columns are ignored when creating a gData object, so they can be left as they are.
## Add genotypes as row names of dropsMap.
rownames(dropsMap) <- dropsMap[["SNP.names"]]
## Rename Chomosome and Position columns.
colnames(dropsMap)[match(c("Chromosome", "Position"), colnames(dropsMap))] <- c("chr", "pos")Now a gData object containing only map and marker information can be created. This object can be used as a base. Phenotypic data, a kinship matrix and covariates may be added later on.
## Create a gData object containing map and marker information.
gDataDrops <- createGData(geno = dropsMarkers, map = dropsMap)The next step involves adding phenotypic data to the gData object
just created. dropsPheno contains genotypic means for 28 trials. To be
able to run a GWAS analysis per trial the data has to be added as a list
of 28 data.frames. These data.frames should have “genotype” as their
first column and may only contain traits after that. Other columns need
to be dropped.
The phenotypic data can be added to gDataDrops using the same
function that was used for creating the object. Adding an argument
gData to the function call will add new data to an existing
gData object. Any phenotypic data already present will be
overwritten.
## Rename Variety_ID to genotype.
colnames(dropsPheno)[colnames(dropsPheno) == "Variety_ID"] <- "genotype"
## Select relevant columns and convert data to a list.
dropsPhenoList <- split(x = dropsPheno[c("genotype", "grain.yield",
"grain.number", "seed.size",
"anthesis", "silking", "plant.height",
"tassel.height", "ear.height")],
f = dropsPheno[["Experiment"]])
## Add phenotypic data to gDataDrops.
gDataDrops <- createGData(gData = gDataDrops, pheno = dropsPhenoList)Note that if the phenotypic data consists of only one trial/experiment, it can be added as a single data.frame without first converting it to a list. In this case createGData will convert the input to a list with one item.
To get an idea of the contents of the data a summary of the gData
object can be made. This will give an overview of the content of the map
and markers and also print a summary per trait per trial. Since there
are 28 trials and 8 traits in gDataDrops giving a very large summary,
the output is restricted here to one trial, Mur13W, using the
trials parameter of the summary function.
## Summarize gDataDrops.
summary(gDataDrops, trials = "Mur13W")
#> map
#> Number of markers: 41722
#> Number of chromosomes: 10
#>
#> markers
#> Number of markers: 41722
#> Number of genotypes: 246
#> Content:
#> 0 1 2 <NA>
#> 0.28 0.01 0.71 0.00
#>
#> pheno
#> Number of trials: 1
#>
#> Mur13W:
#> Number of traits: 8
#> Number of genotypes: 246
#>
#> grain.yield grain.number seed.size anthesis silking plant.height tassel.height ear.height
#> Min. 3.3 1348 229 56 59 222 NA 102
#> 1st Qu. 6.3 2641 284 61 64 251 NA 125
#> Median 7.5 2965 298 63 66 258 NA 133
#> Mean 7.4 2986 297 63 66 259 NaN 133
#> 3rd Qu. 8.4 3359 312 66 68 266 NA 141
#> Max. 11.4 4510 365 71 74 294 NA 172
#> NA's 0.0 0 0 0 0 0 246 0It is also possible to plot the genetic map for a gData object. This will show the length of the chromosomes and the position of the markers in the gData object on the chromosomes. It is also possible to highlight one or more markers.
## Plot genetic map.
plot(gDataDrops)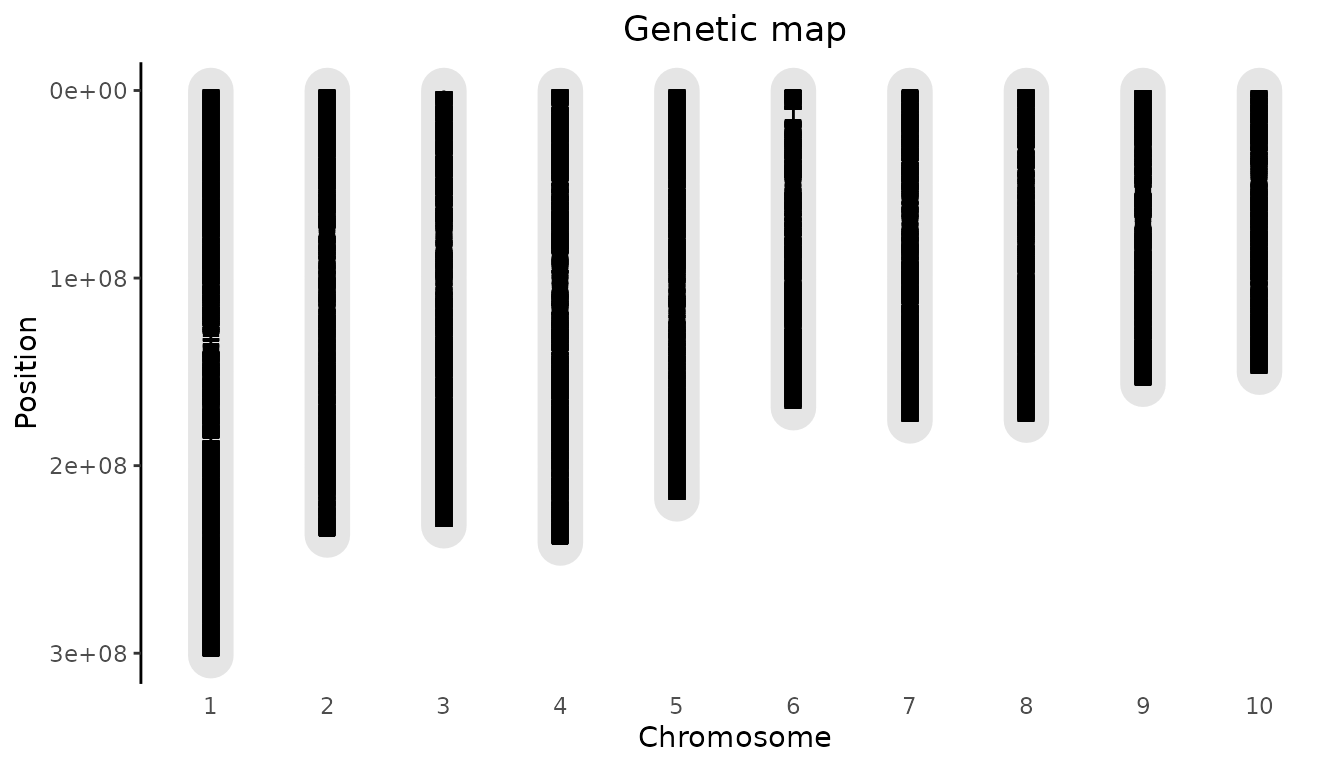
To highlight a marker, the parameter highlight can be
used. It requires data.frame with at least the column “chr”
and “pos”. If a column name is present it will be used to annotate the
highlighted marker. If no name column is present the annotation will be
of the form pos@chr.
## Plot genetic map.
## Highlight the 20.000th marker in the map.
plot(gDataDrops, highlight = dropsMap[20000, ])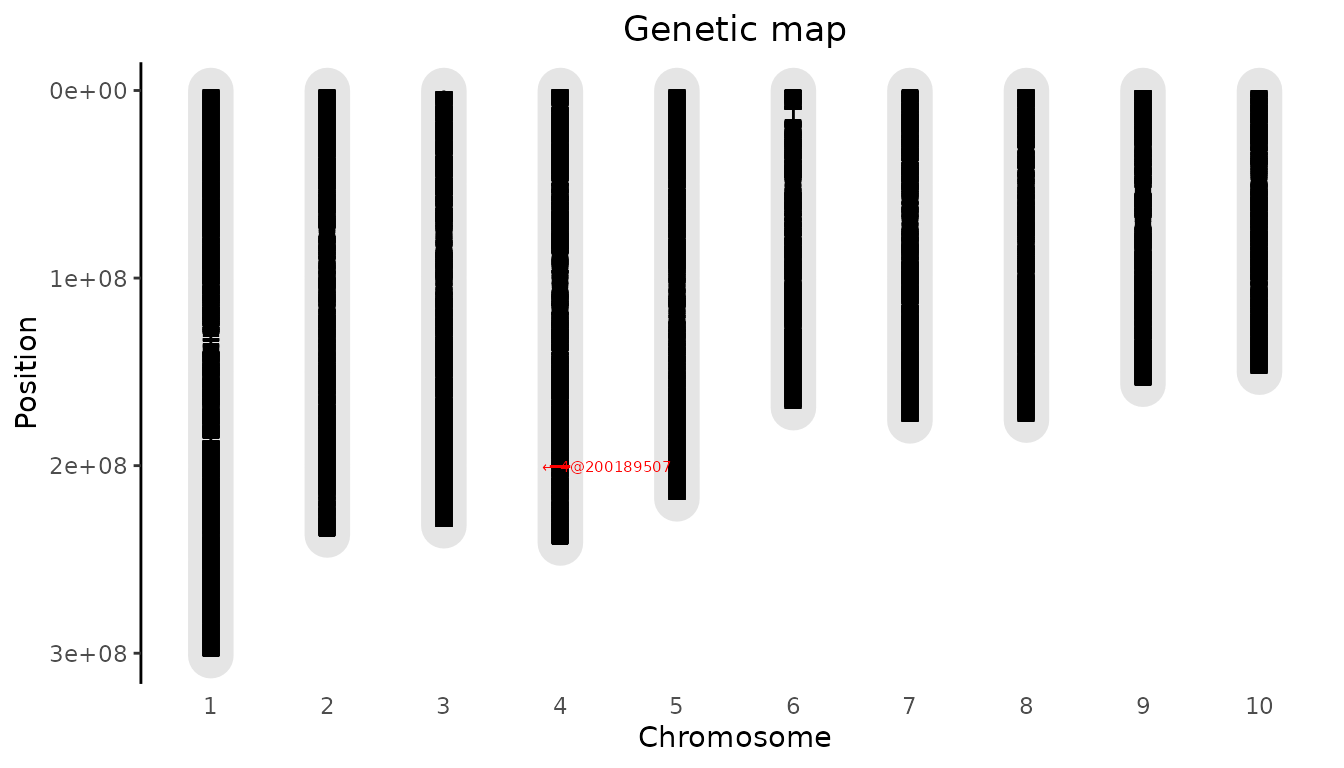
Recoding and cleaning of markers
Marker data has to be numerical and without missing values in order to do GWAS analysis. This can be achieved using the codeMarkers function. The marker data available for the DROPS project has already been converted from A/T/C/G to 0/1/2. The codeMarkers function in the package can still be used to further clean the markers, e.g. by removing the duplicate SNPs.
## Remove duplicate SNPs from gDataDrops.
gDataDropsDedup <- codeMarkers(gDataDrops, impute = FALSE, verbose = TRUE)
#> Input contains 41722 SNPs for 246 genotypes.
#> 0 genotypes removed because proportion of missing values larger than or equal to 1.
#> 0 SNPs removed because proportion of missing values larger than or equal to 1.
#> 5098 duplicate SNPs removed.
#> Output contains 36624 SNPs for 246 genotypes.Note that in case of duplicate SNPs only one of them is used - this is picked at random. To get reproducible results ake sure to set a seed for the random number generator.
Since missing values are not uncommon in real data, the codeMarkers function has several imputation options. To demonstrate these, we randomly replace 1% of the values in the marker matrix by NA.
## Copy gData object.
gDataDropsMiss <- gDataDrops
## Add random missing values to 1% of the values in the marker matrix.
set.seed(1)
nVal <- nrow(gDataDropsMiss$markers) * ncol(gDataDropsMiss$markers)
gDataDropsMiss$markers[sample(x = 1:nVal, size = nVal / 100)] <- NATo impute the missing values added above, codeMarkers has to
be run with impute = TRUE. Then the type of imputation can
be chosen setting the parameter imputeType:
-
imputeType = "fixed": Impute all missing values by a single fixed value. Use the parameterfixedValueto set this value. -
imputeType = "random": Impute missing values with a random value based on the non-missing values for a SNP. -
imputeType = "beagle": Impute missing values using the independent beagle software (Browning and Browning 2007). Version 4.1 of this software is included in the package and is called with its default settings. It requires java to run. Since running beagle takes a long time for the current data, the call to beagle is only shown here but not run.
Before imputation, genotypes with a high proportion of missing values
in the marker matrix can be removed by setting the parameter nMissGeno.
Setting it to 0.1 will remove all genotypes with a proportion of missing
values higher than 0.1. In a similar way SNPs with a high proportion of
missing values can be removed setting nMiss. The default value for both
parameters is 1, so genotypes and SNPs with only missing values are
always removed.
The parameter MAF can be used to remove SNPs with a Minor Allele
Frequency (MAF) lower than the specified MAF. After imputation, the
markers are again scanned for duplicate SNPs and SNPs with a MAF lower
than the threshold - if these are found, they will be removed as
described earlier.
## Impute missing values with random value.
## Remove SNPs and genotypes with proportion of NA larger than 0.01.
gDataDropsImputed <- codeMarkers(gData = gDataDropsMiss,
nMissGeno = 0.01,
nMiss = 0.01,
impute = TRUE,
imputeType = "random",
verbose = TRUE)
#> Input contains 41722 SNPs for 246 genotypes.
#> 119 genotypes removed because proportion of missing values larger than or equal to 0.01.
#> 14341 SNPs removed because proportion of missing values larger than or equal to 0.01.
#> 882 duplicate SNPs removed.
#> 15213 missing values imputed.
#> 1046 duplicate SNPs removed after imputation.
#> Output contains 25453 SNPs for 127 genotypes.
## Impute missing values using beagle software.
gDataDropsImputedBeagle <- codeMarkers(gData = gDataDropsMiss,
impute = TRUE,
imputeType = "beagle",
verbose = TRUE)Single trait GWAS
The cleaned gData object can be used for performing single trait GWAS analysis with the runSingleTraitGwas function. The default parameter values in the function are chosen in such a way that usually the analysis gives decent results. In the examples below the trial Mur13W is used to demonstrate the options of the runSingleTraitGwas function.
## Run single trait GWAS for traits 'grain.yield' and 'anthesis' for trial Mur13W.
GWASDrops <- runSingleTraitGwas(gData = gDataDropsDedup,
trials = "Mur13W",
traits = c("grain.yield", "anthesis"))The output of the runSingleTraitGwas function is an object
of class GWAS. This is a list consisting of five components described
below.
GWAResult: a list of data.tables, one for each trial
for which the analysis was run. Each data.table has the following
columns:
| trait | trait name |
| snp | SNP name |
| chr | chromosome on which the SNP is located |
| pos | position of the SNP on the chromosome |
| allFreq | allele frequency of the SNP |
| pValue | P-value for the SNP |
| effect | effect of the SNP on the trait value |
| effectSe | standard error of the effect of the SNP on the trait value |
| RLR2 | likelihood-ratio-based R2 as defined in Sun et al. (2010) |
| LOD | LOD score for the SNP, defined as -\log_{10}(pValue) |
print(head(GWASDrops$GWAResult$Mur13W), row.names = FALSE)
#> trait snp chr pos allFreq pValue effect effectSe RLR2 LOD
#> <char> <char> <int> <int> <num> <num> <num> <num> <num> <num>
#> grain.yield SYN83 1 3498 0.60 0.38 -0.084 0.097 0.00310 0.417
#> grain.yield PZE-101000060 1 157104 0.72 0.51 0.067 0.102 0.00176 0.291
#> grain.yield PZE-101000088 1 238347 0.84 0.83 -0.027 0.125 0.00019 0.081
#> grain.yield PZE-101000083 1 239225 0.58 0.22 -0.113 0.092 0.00615 0.660
#> grain.yield PZE-101000108 1 255850 0.90 0.47 -0.124 0.170 0.00215 0.330
#> grain.yield PZE-101000111 1 263938 0.83 0.32 -0.117 0.119 0.00395 0.489Note that the estimated effect is computed for a single allele. Its direction depends on the coding of the markers in the gData object. In this example the minor allele was used as reference allele, so the effects are the estimated effects for the minor allele.
signSnp: a list of data.tables, one for each trial for which the analysis was run, containing the significant SNPs. Optionally also the SNPs close to the significant SNPs are included in the data.table. See 3.6.7 for more information on how to do this. The data.tables in signSnp consist of the same columns as those in GWAResult described above. Two extra columns are added:
| snpStatus | either “significant SNP” or “within … of a significant SNP” |
| propSnpVar | proportion of the variance explained by the SNP, computed as \beta_{\textrm{SNP}}^2 * var(\textrm{SNP}) / var(\textrm{pheno}) |
print(GWASDrops$signSnp$Mur13W, row.names = FALSE)
#> trait snp chr pos allFreq pValue effect effectSe RLR2 LOD
#> <char> <char> <int> <int> <num> <num> <num> <num> <num> <num>
#> grain.yield PZE-106021363 6 18846283 0.70 3.5e-07 0.52 0.1 0.096 6.5
#> grain.yield PZE-106021410 6 18990291 0.70 2.3e-07 0.53 0.1 0.098 6.6
#> grain.yield PZE-106021419 6 18991091 0.74 2.9e-07 0.53 0.1 0.097 6.5
#> grain.yield PZE-106021420 6 18991117 0.70 1.3e-06 0.50 0.1 0.087 5.9
#> grain.yield PZE-106021424 6 18991481 0.74 5.0e-07 0.52 0.1 0.093 6.3
#> snpStatus propSnpVar
#> <fctr> <num>
#> significant SNP 0.088
#> significant SNP 0.090
#> significant SNP 0.083
#> significant SNP 0.080
#> significant SNP 0.080kinship: the kinship matrix (or matrices) used in the GWAS analysis. This can either be the user provided kinship matrix or the kinship matrix computed when running the runSingleTraitGwas function.
thr: a list of thresholds, one for each trial for which the analysis was run, used for determining significant SNPs.
GWASInfo: additional information on the analysis, e.g. the call and the type of threshold used.
GWAS Summary
For a quick overview of the results, e.g. the number of significant SNPs, use the summary function.
## Create summary of GWASDrops.
summary(GWASDrops)
#> Mur13W:
#> Traits analysed: grain.yield, anthesis
#>
#> Data are available for 36624 SNPs.
#> 0 of them were not analyzed because their minor allele frequency is below 0.01
#>
#> GLSMethod: single
#> kinshipMethod: astle
#>
#> Trait: grain.yield
#>
#> Mixed model with only polygenic effects, and no marker effects:
#> Genetic variance: 0.96
#> Residual variance: 0.21
#>
#> LOD-threshold: 5.9
#> Number of significant SNPs: 5
#> Smallest p-value among the significant SNPs: 2.3e-07
#> Largest p-value among the significant SNPs: 1.3e-06 (LOD-score: 5.9)
#>
#> No genomic control correction was applied
#> Genomic control inflation-factor: 0.99
#>
#> Trait: anthesis
#>
#> Mixed model with only polygenic effects, and no marker effects:
#> Genetic variance: 4
#> Residual variance: 0.87
#>
#> LOD-threshold: 5.9
#> Number of significant SNPs: 0
#>
#> No genomic control correction was applied
#> Genomic control inflation-factor: 0.97GWAS Plots
The plot.GWAS function can be used to visualize the results in GWASDrops, with a QQ-plot, Manhattan plot or QTL-plot.
QQ-plots
A QQ-plot of the observed against the expected -\log_{10}(p) values can be made by setting
plotType = "qq". Most of the SNPs are expected to have no
effect, resulting in P-values uniformly distributed on [0,1], and leading to the identity function
(y=x) on the -\log_{10}(p) scale. As in the plot below,
deviations from this line should only occur on the right side of the
plot, for a small number of SNPs with an effect on the phenotype (and
possibly SNPs in LD). There is inflation if the
observed -\log_{10}(p) values are
always above the line y=x, and (less
common) deflation if they are always below this line. A
QQ-plot therefore gives a first impression of the quality of the GWAS
model: if for example -\log_{10}(p)
values are consistently too large (inflation), the correction for
genetic relatedness may not be adequate.
## Plot a QQ-plot of GWAS Drops.
plot(GWASDrops, plotType = "qq", trait = "grain.yield")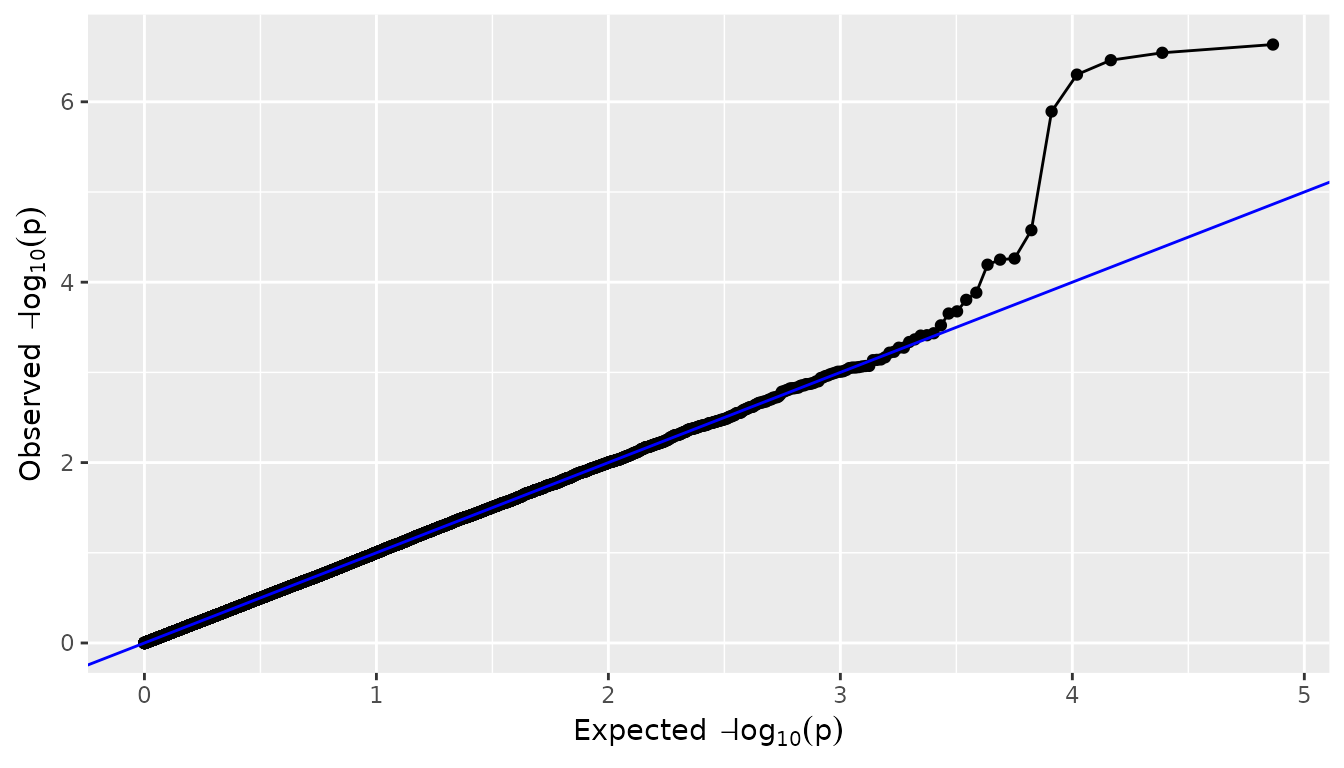
Manhattan plots
A manhattan plot is made by setting
plotType = "manhattan". Significant SNPs are marked in
red.
## Plot a manhattan plot of GWAS Drops.
plot(GWASDrops, plotType = "manhattan", trait = "grain.yield")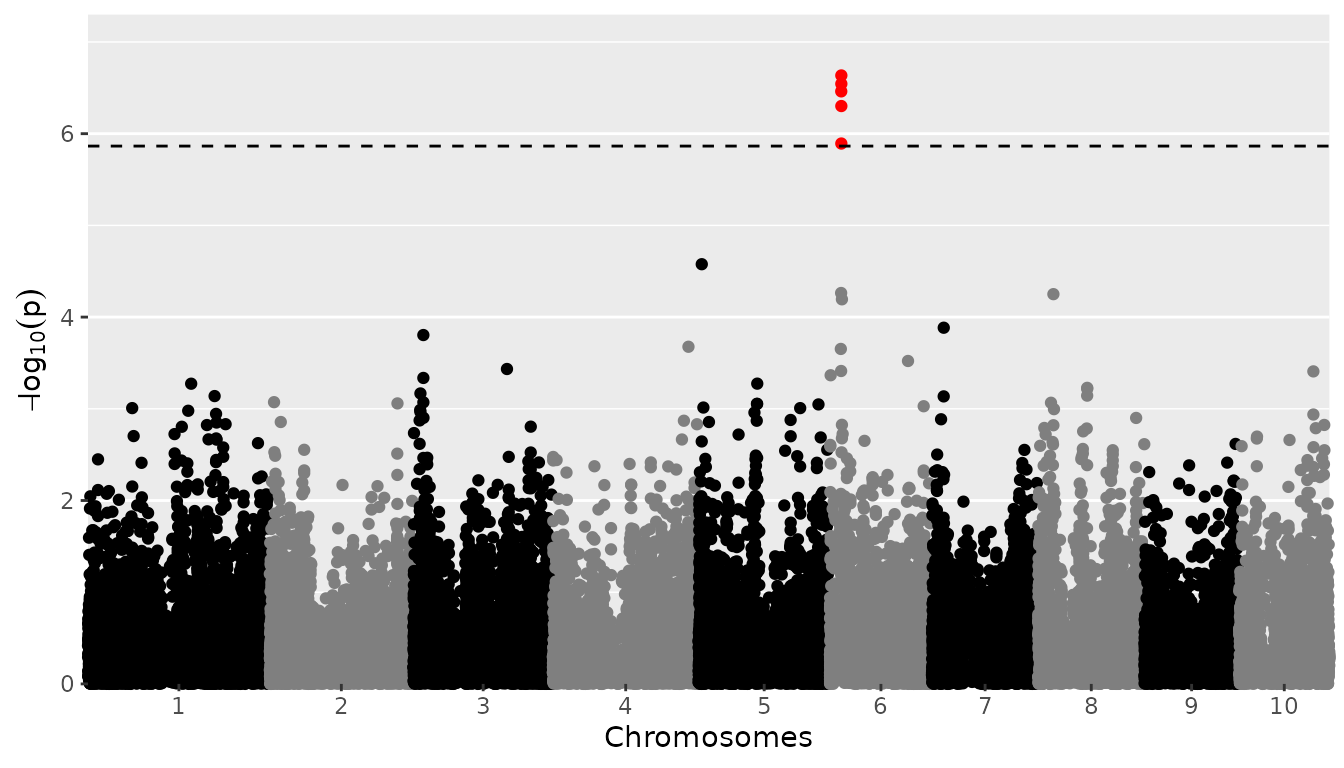
To change the threshold when plotting, without having to redo the
GWAS analysis, use the parameter yThr. Use chr
to plot a subset of chromosomes.
## Plot a manhattan plot of GWAS Drops.
## Set significance threshold to 4 and only plot chromosomes 6 to 8.
plot(GWASDrops, plotType = "manhattan", trait = "grain.yield", yThr = 4, chr = 6:8)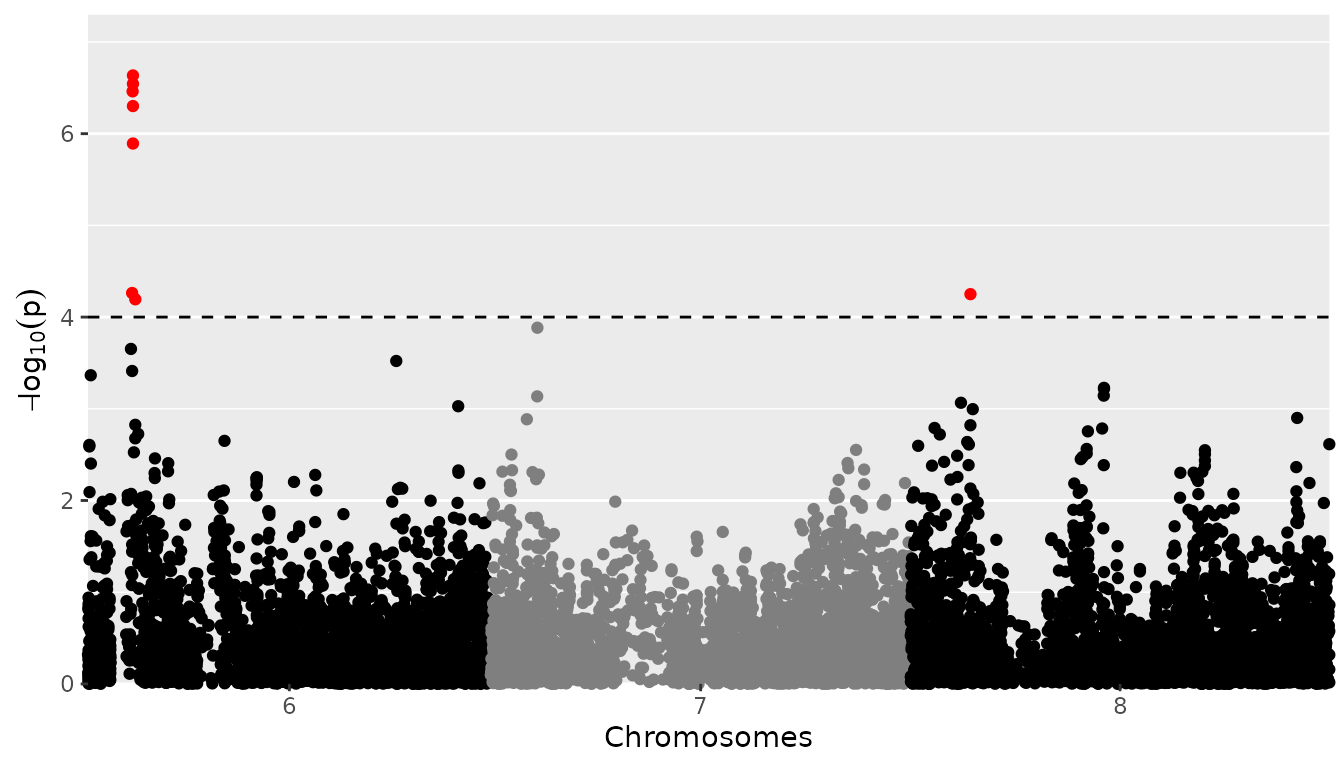 When plotting a single chromosome it is possible to specify
When plotting a single chromosome it is possible to specify
startPos and/or endPos to indicate the start
position and end position for SNPs that are plotted.
## Plot a manhattan plot of GWAS Drops.
## Set significance threshold to 4 and only plot first part of chromosome 6.
plot(GWASDrops, plotType = "manhattan", trait = "grain.yield",
yThr = 4, chr = 6, startPos = 0, endPos = 6e7)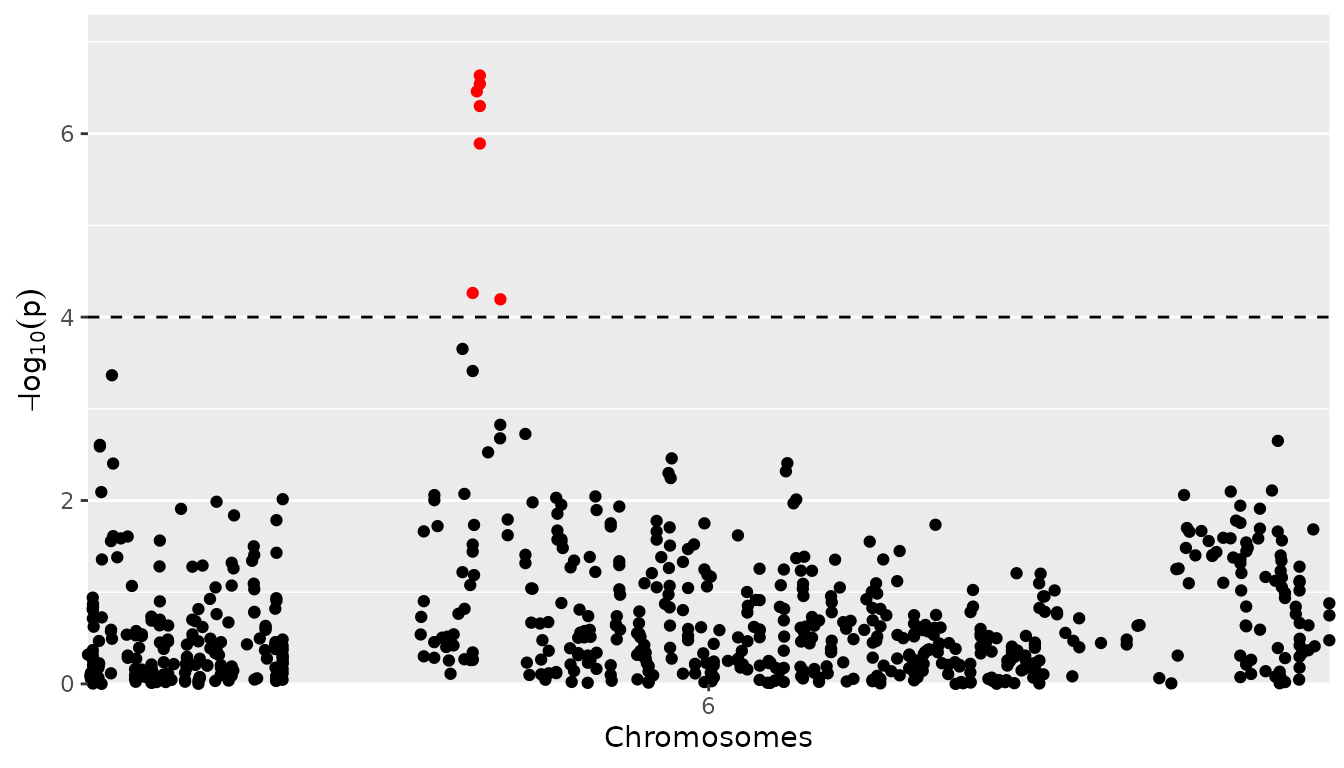
If the number of SNPs gets large, the plotting might get slow. This
can be dealt with by setting the parameter lod. When doing
so, for the SNPs with a LOD-value below lod, only 5% will
be plotted. The chance of a SNP being plotted is proportional to its
LOD-value. SNPs with a higher LOD have a higher chance of being plotted.
Since most SNPs will have low LOD-values, this restriction doesn’t very
much affect the resulting plot. Note that the 5% of the SNPs that is
plotted, is selected randomly. For reproducible results make sure to set
a seed.
## Plot a manhattan plot of GWAS Drops.
## Plot only 5% of SNPs with a LOD below 3.
set.seed(1)
plot(GWASDrops, plotType = "manhattan", trait = "grain.yield", lod = 3)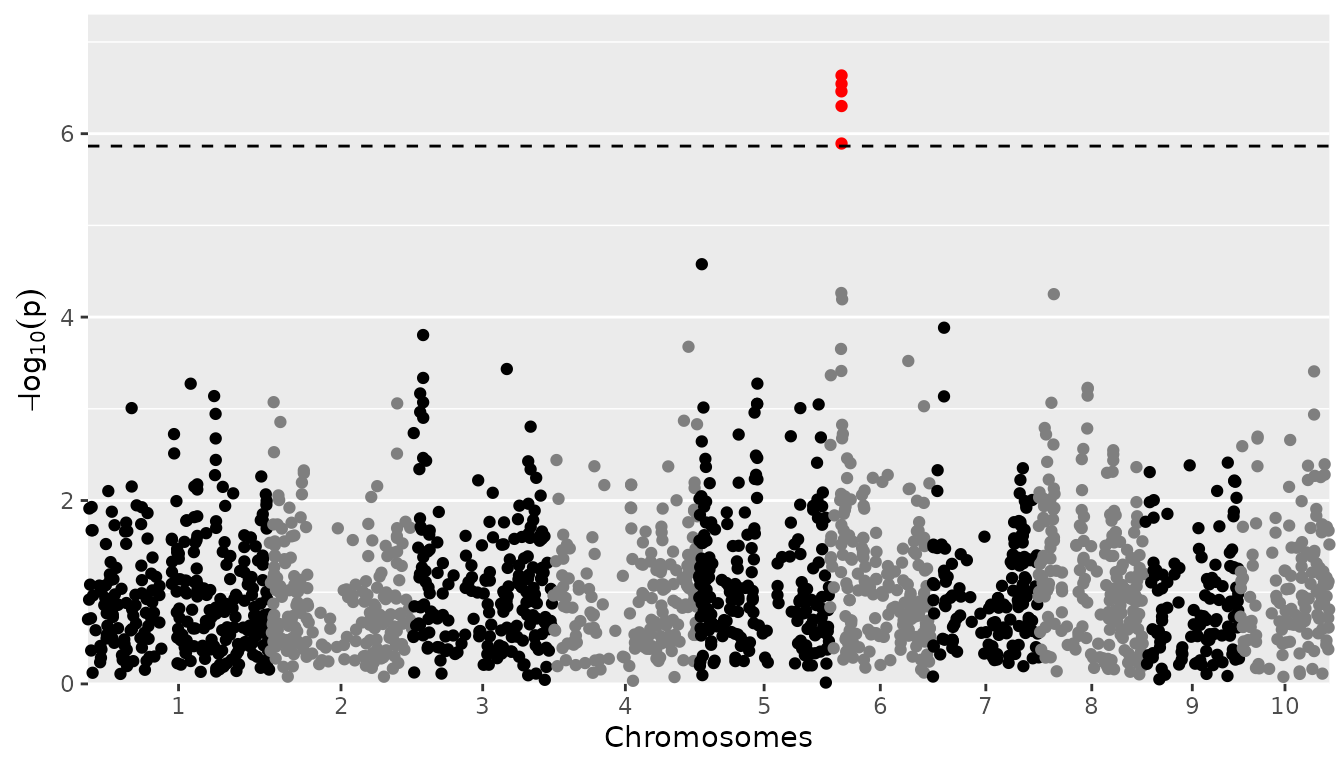
The user can specify known effects (effects), which will
be used for determining true/false positives and false negatives. These
are displayed in green, orange and yellow, respectively.
## Plot a manhattan plot of GWAS Drops with significance threshold 4.
## Assume PZE-106021410 and PZE-105012420 are SNPs with known effects.
plot(GWASDrops, plotType = "manhattan", trait = "grain.yield",
effects = c("PZE-106021410", "PZE-105012420"))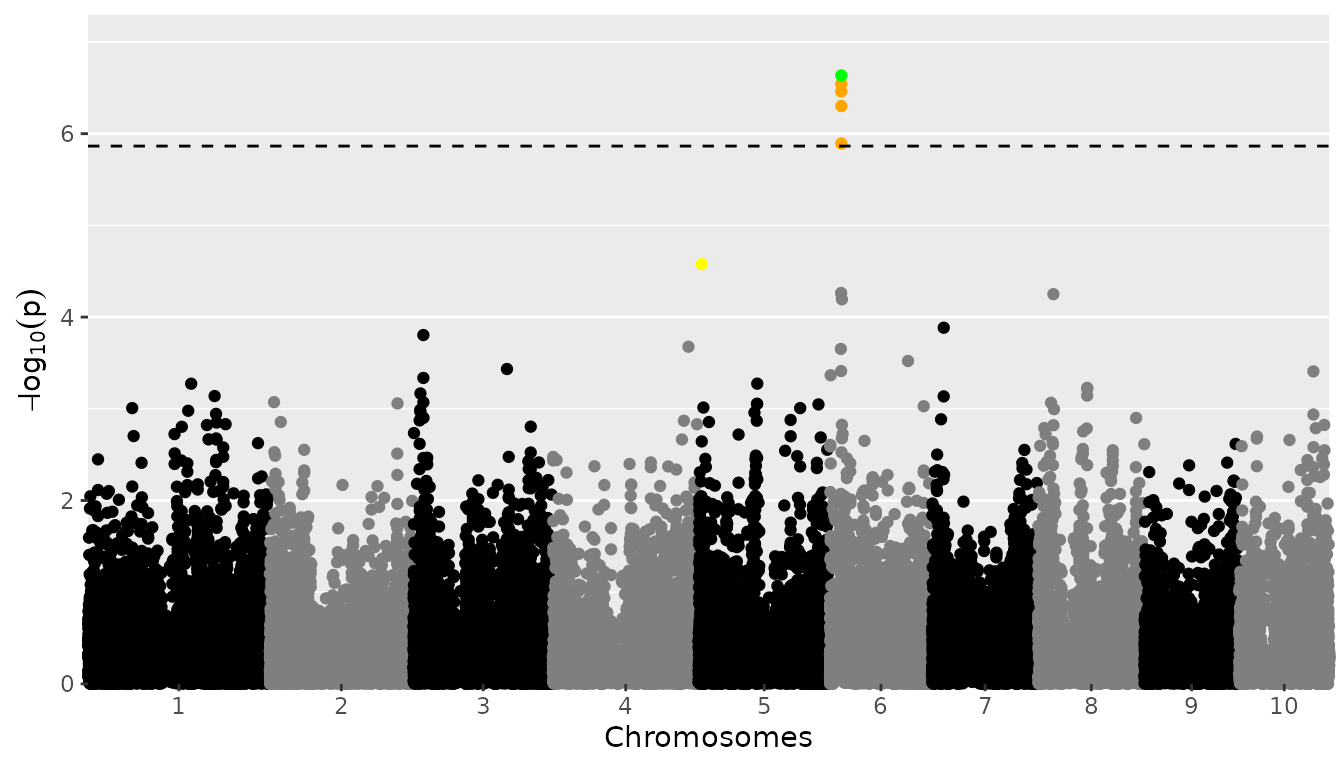 The plot now shows PZE-106021410 in green, since it is a SNP with a
known effect also found by our GWAS analyses. The other SNPs on
chromosome 6 are shown in orange, they are found by our GWAS, but not
known effects. PZE-105012420 is shown in yellow, it is a SNP that has a
known effect but it isn’t picked up as significant by our analysis. Note
that PZE-106021410 and PZE-105012420 are chosen solely to demonstrate
how the effects options works when making a manhattan plot, they don’t
represent truly known effects.
The plot now shows PZE-106021410 in green, since it is a SNP with a
known effect also found by our GWAS analyses. The other SNPs on
chromosome 6 are shown in orange, they are found by our GWAS, but not
known effects. PZE-105012420 is shown in yellow, it is a SNP that has a
known effect but it isn’t picked up as significant by our analysis. Note
that PZE-106021410 and PZE-105012420 are chosen solely to demonstrate
how the effects options works when making a manhattan plot, they don’t
represent truly known effects.
Also the visual appearance of the plot can be altered, e.g by
specifying the colors for the chromosomes or a custom title. See for
specifics on how to do this help(plot.GWAS).
QTL plots
A qtl plot can be made by setting plotType = "qtl". In
this plot the significant SNPs are marked by circles at their genomic
positions, with diameter proportional to the estimated effect size; for
an example see E. Millet et al. (2016). Typically, this is done for
multiple traits or environments, with the genomic position on the
x-axis, which are displayed horizontally above each other and can thus
be compared.
## Plot a qtl plot of GWAS Drops for Mur13W.
plot(GWASDrops, plotType = "qtl")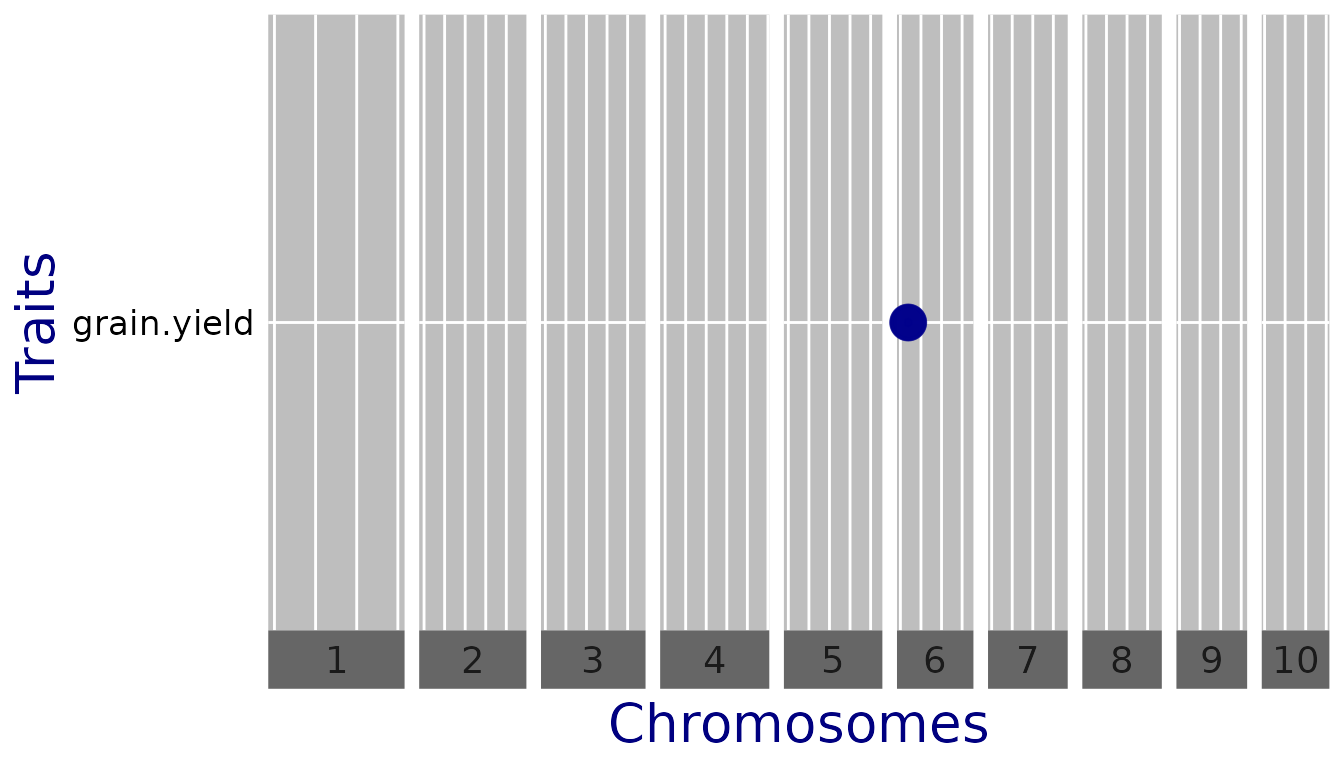
In this case, for “anthesis” there are no significant SNPs so the
line is left out. To change the threshold when plotting without having
to redo the GWAS analysis, use the parameter yThr.
## Plot a qtl plot of GWAS Drops for Mur13W.
## Set significance threshold to 4.
plot(GWASDrops, plotType = "qtl", yThr = 4)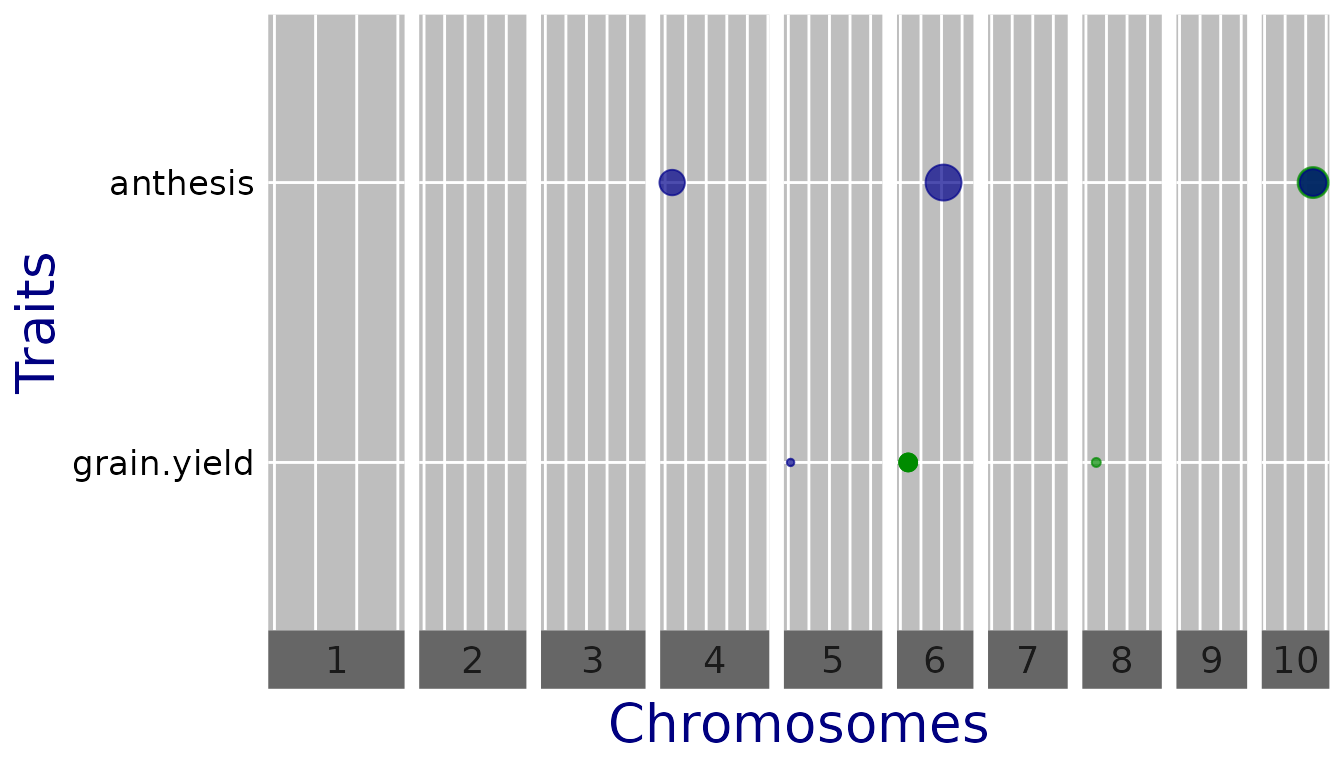
Now there are significant SNPs for both traits. However since the
traits are measured on a different scale, the effect estimates cannot be
compared. For better comparison, one can set
normalize = TRUE, which divides the estimates by the
standard deviation of the phenotype.
## Plot a qtl plot of GWAS Drops for Mur13W.
## Set significance threshold to 4 and normalize effect estimates.
plot(GWASDrops, plotType = "qtl", yThr = 4, normalize = TRUE)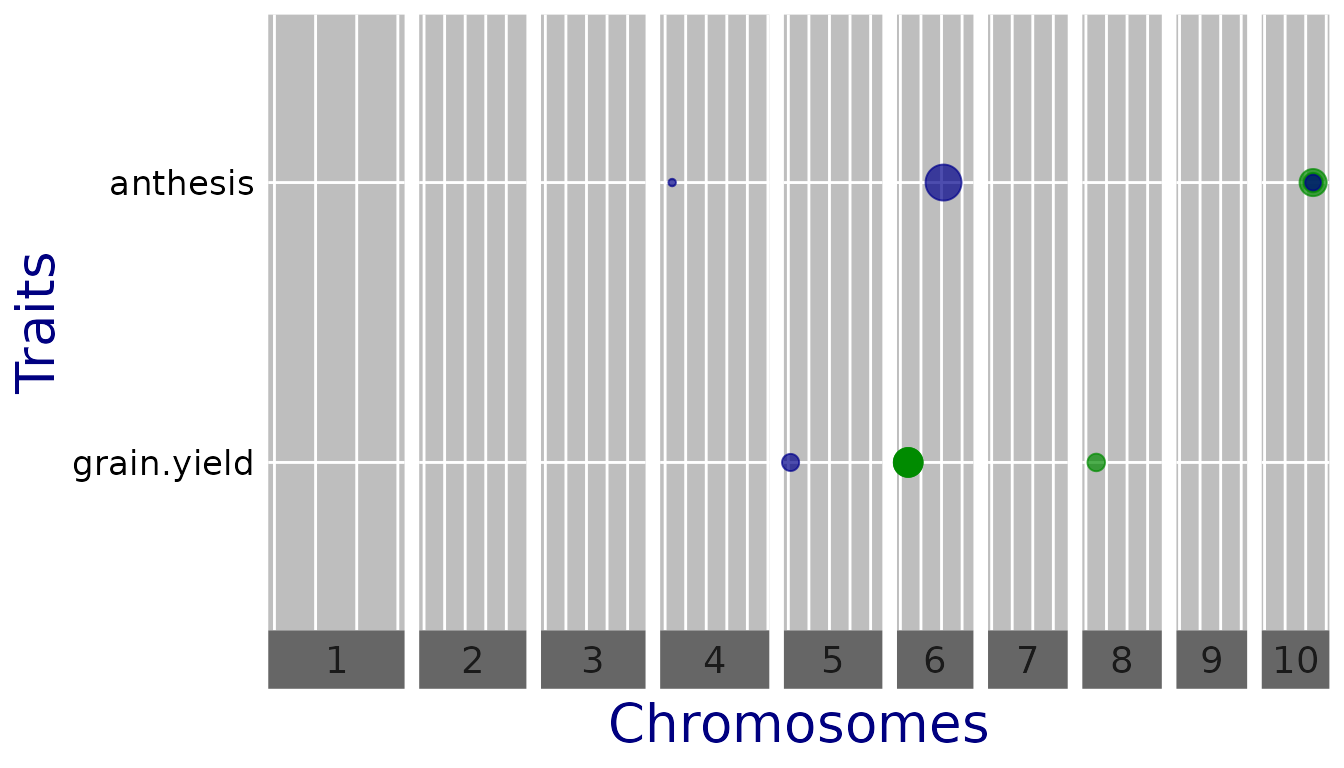
Now the effects are on the same scale and comparable.
Other parameters can be used to plot a subset of the chromosomes
(chr) and directly export the plot to .pptx
(exportPptx = TRUE and specify pptxName). Note
that the officer package is required for this. A full list of parameters
can be found by running help(plot.GWAS).
Kinship matrices
The runSingleTraitGwas function has an argument
kinshipMethod, which defines the kinship matrix used for
association mapping. Kinship matrices can be computed directly using the
kinship function or within the runSingleTraitGwas
function. There are four options:
- using the covariance between the scaled SNP-scores
(
kinshipMethod = "astle", the default; see e.g. equation (2.2) in Astle and Balding (2009)) - Identity by State (
kinshipMethod = "IBS"; see e.g. equation (2.3) in Astle and Balding (2009)) - using the formula by VanRaden (2008)
(
kinshipMethod = "vanRaden") - User-defined, in which case the parameter
kinneeds to be specified.
By default, the same kinship matrix is used for testing all SNPs
(GLSMethod = "single"). When
GLSMethod = "multi", the kinship matrix is
chromosome-specific. In this case, the function fits variance components
and computes effect-estimates and P-values for each chromosome in turn,
using the kinship matrix for that chromosome. Each chromosome-specific
kinship matrix is computed using the method specified by the argument
kinshipMethod, using all SNPs that are not
on this chromosome. As shown by Rincent et al.
(2014), this often gives a
considerable improvement in power.
## Run single trait GWAS for trial 'Mur13W' and trait 'grain.yield'
## Use chromosome specific kinship matrices computed using method of van Raden.
GWASDropsChrSpec <- runSingleTraitGwas(gData = gDataDropsDedup,
traits = "grain.yield",
trials = "Mur13W",
GLSMethod = "multi",
kinshipMethod = "vanRaden")Further options
The runSingleTraitGwas function has many more parameters that can be specified. In this section similar parameters are grouped and explained with examples on how to use them.
Multiple trials and traits
The analysis can be performed for more than one trait in a single
function call. Not specifying traits at all will perform
the analysis for all traits that are present in the phenotypic data in
the gData object. The results will be combined in a single output object
with the traits specified in the column trait in
GWAResult and signSnp within this object. Running for
multiple trials is also possible and will generate a list of outputs
within GWAResult and signSnp in the output object. Not
specifying trials will run the GWAS analysis for all trials
in the phenotypic data in the gData object.
Significance thresholds
The threshold for selecting significant SNPs in a GWAS analysis is
computed by default using Bonferroni correction, with an alpha of 0.05.
The alpha can be modified setting the option alpha when calling
runSingleTraitGwas. Two other threshold types can be used: a
fixed threshold (thrType = "fixed") specifying the -\log_{10}(p) (LODThr) value of the
threshold, or a threshold that defines the n SNPs with the highest -\log_{10}(p) scores as significant SNPs. Set
thrType = "small" together with nSnpLOD = n to
do this. In the following example, we select all SNPs with p < 10^{-4}.
## Run single trait GWAS for trait 'grain.yield' for Mur13W.
## Use a fixed significance threshold of 4.
GWASDropsFixThr <- runSingleTraitGwas(gData = gDataDropsDedup,
trials = "Mur13W",
traits = "grain.yield",
thrType = "fixed",
LODThr = 4)Controlling false discovery rate
A final option for selecting significant SNPs is by setting
thrType = "fdr". When doing so the significant SNPs won’t
be selected by computing a genome wide threshold, but by trying to
control the rate of false discoveries as in Brzyski et al. (2016).
First, a list is defined containing all SNPs with a p-Value below
pThr. Then clusters of SNPs are created using a two step
iterative process in which SNPs with the lowest p-values are selected as
cluster representatives. This SNP and all SNPs that have a correlation
with this SNP of \rho or higher
(specified by the function parameter rho) will form a
cluster. The selected SNPs are removed from the list and the procedure
is repeated until no SNPs are left. At the end of this step, one has a
list of clusters, with corresponding vector of p-values of the cluster
representatives. Finally, to determine the number of significant
clusters, the first cluster is determined for which the p-value of the
cluster representative is larger than cluster_{number} * \alpha / m, where m is the number of SNPs and \alpha can be specified by the corresponding
function parameter. All previous clusters are selected as
significant.
Note that because of the way the significant SNPs are selected it is
possible that a SNP with a lower p-Value than a significant SNP is not
significant itself. This happens if that SNP is highly correlated to
another SNP with an even lower p-Value. Because of this it is not
possible to determine a threshold for the p-Value below which SNPs are
significant. The value for the threshold in the output will therefor be
NA and no line will be plotted for it in the manhattan
plot.
Variance covariance matrix
There are two ways to compute the phenotypic variance covariance
matrix used in the GWAS analysis. Either the EMMA algorithm or the
Newton-Raphson algorithm. Specify the method by setting the parameter
remlAlgo to either "EMMA" or
"NR". By default the EMMA algorithm is used.
## Run single trait GWAS for trait 'grain.yield' for Mur13W.
## Use the Newton Raphson algorithm for computing the variance components.
GWASDropsNR <- runSingleTraitGwas(gData = gDataDropsDedup,
trials = "Mur13W",
traits = "grain.yield",
remlAlgo = "NR")Genomic control correction
In the GWAS object returned by runSingleTraitGwas, the inflation factor (Devlin and Roeder 1999) is returned in the GWASInfo. It is also shown in the summary. Ideally this factor should be 1, meaning there is no inflation at all. In practice however this rarely happens. Its value for Mur13W can be checked as follows:
GWASDrops$GWASInfo$inflationFactor$Mur13W
#> grain.yield anthesis
#> 0.99 0.97Both values are very close to 1, so in this case there is no need to
apply any correction based on this. If the values are further away from
1, the inflation can be corrected for by setting
genomicControl = TRUE in runSingleTraitGwas.
## Run single trait GWAS for trait 'grain.yield' for Mur13W.
## Perform genomic correction on the p-Values.
GWASDropsGenControl <- runSingleTraitGwas(gData = gDataDropsDedup,
trials = "Mur13W",
traits = "grain.yield",
genomicControl = TRUE)Covariates
Covariates can be included as extra fixed effects in the GWAS model.
The runSingleTraitGwas function distinguishes between ‘usual’
covariates and SNP-covariates. The former could be design factors such
as block, or other traits one wants to condition on. In the latter case,
the covariate(s) are one or more of the markers contained in the
genotypic data. SNP-covariates can be set with the parameter
snpCov, which should be a vector of marker-names.
Similarly, other covariates should be specified using the parameter
covar, containing a vector of covariate names. The gData
object should contain these covariates in gData$covar.
In case SNP-covariates are used, GWAS for all the other SNPs is performed with the the SNP-covariates as extra fixed effect; also the null model used to estimate the variance components includes these effects. For each SNP in SNP-covariates, a p-value is obtained using the same F-test and null model to estimate the variance components, but with only all other SNPs (if any) in SNP-covariates as fixed effects.
## Run single trait GWAS for trait 'grain.yield' for Mur13W.
## Use PZE-106021410, the most significant SNP, a SNP covariate.
GWASDropsSnpCov <- runSingleTraitGwas(gData = gDataDropsDedup,
trials = "Mur13W",
traits = "grain.yield",
snpCov = "PZE-106021410")Minor Allele Frequency
It is recommended to remove SNPs with a low minor allele frequency
(MAF) from the data before starting a GWAS analysis. However it is also
possible to do so in the analysis itself. The difference between these
approaches is that codeMarkers removes the SNPs, whereas
runSingleTraitGwas excludes them from the analysis but leaves
them in the output (with results set to NA). In the latter case it will
still be possible to see the allele frequency of the SNP.
By default all SNPs with a MAF lower than 0.01 are excluded from the
analysis. This can be controlled by the parameter MAF.
Setting MAF to 0 will still exclude duplicate SNPs since duplicates
cause problems when fitting the underlying models. Instead of excluding
SNPs with a low MAF it is also possible to exclude SNPs with a low minor
allele count (MAC). This is done setting useMAF = FALSE and
then specifying MAC.
Note that the computation of allele frequencies in
runSingleTraitGwas is done after the removal of genotypes that
have missing values for the trait for which the analysis is done. This
could lead to slightly different values for the allele frequencies than
when these are computed in codeMarkers (which only uses the
genotypic data) resulting in the removal a slightly different set of
markers.
## Run single trait GWAS for trait 'grain.yield' for Mur13W.
## Only include SNPs that have a MAC of at least 20
GWASDropsMAC <- runSingleTraitGwas(gData = gDataDropsDedup,
trials = "Mur13W",
traits = "grain.yield",
useMAF = FALSE,
MAC = 20)SNPs close to significant SNPs
Apart from the significant SNPs themselves, it is usually of interest
to identify SNPs that are close to the significant SNPs. These can be
included in the output by setting sizeIncludedRegion to a
positive value. When doing so, SNPs that are within a distance of
sizeIncludedRegion from a significant SNP (based on the
information in the map) are included in the signSnp data.table in the
output. Only SNPs having sufficient linkage disequilibrium (LD) with the
significant SNP are included. LD is measured in terms of the squared
Pearson correlation (R^2). The minimum
R^2 required to be included can be
specified by setting minR2.
## Run single trait GWAS for trait 'grain.yield' for Mur13W.
## Include SNPs within 200000 centimorgan of significant SNPs with a minimum LD of 0.1.
GWASDropsInclClose <- runSingleTraitGwas(gData = gDataDropsDedup,
trials = "Mur13W",
traits = "grain.yield",
sizeInclRegion = 200000,
minR2 = 0.1)
## Check signSnp in output.
print(head(GWASDropsInclClose$signSnp$Mur13W), row.names = FALSE)
#> trait snp chr pos allFreq pValue effect effectSe RLR2 LOD
#> <char> <char> <int> <int> <num> <num> <num> <num> <num> <num>
#> grain.yield SYN25279 6 18646353 0.76 3.9e-04 0.39 0.11 0.049 3.4
#> grain.yield SYN25281 6 18646369 0.79 5.5e-05 0.46 0.11 0.062 4.3
#> grain.yield SYN25282 6 18646737 0.67 3.0e-02 0.24 0.11 0.019 1.5
#> grain.yield SYN22470 6 18712443 0.62 1.8e-02 0.25 0.11 0.022 1.7
#> grain.yield PZE-106021363 6 18846283 0.70 3.5e-07 0.52 0.10 0.096 6.5
#> grain.yield PZE-106021410 6 18990291 0.70 2.3e-07 0.53 0.10 0.098 6.6
#> snpStatus propSnpVar
#> <fctr> <num>
#> within 2e+05 of a significant SNP 0.043
#> within 2e+05 of a significant SNP 0.054
#> within 2e+05 of a significant SNP 0.019
#> within 2e+05 of a significant SNP 0.023
#> significant SNP 0.088
#> significant SNP 0.090